Research
Data-driven materials discovery, design and understanding:
Our group is looking for new and imporved materials and a better chemical understanding based on high-throughput computations (mostly density functional theory) and data analysis.
Chemical Heuristics
During her postdoc, Janine has worked on testing and developing chemical heuristics (intuitive rules, usually based on limited data) to further the understanding in solid-state chemistry and physics. For example, she has assessed the predictive power of the Pauling rules on the stability of crystal structures. To do so, an automatic tool for the determination of coordination environments has been developed as well. Together with Geoffroy Hautier, she outlined a vision for the development in the field.
- J. George, G. Hautier, “The chemist versus the machine: traditional knowledge versus machine learning techniques”, Trends in Chemistry, 2021, 3, 86-95.
- D. Waroquiers, J. George, M. Horton, S. Schenk, K. Persson, G.-M. Rignanese, X. Gonze, G. Hautier, “ChemEnv: A Fast and Robust Coordination Environment, Identification Tool”, Acta Cryst. B 2020, 76, 683.
- J. George, D. Waroquiers, D. Di Stefano, G. Petretto, G.-M. Rignanese, G. Hautier, “The Limited Predictive Power of the Pauling Rules”, Angew. Chem. Int. Ed. 2020, 59, 7569
High-throughput searches
I have implemented tools in Python Library pymatgen that allow the automated bonding analysis of inorganic materials with the program Lobster. These tools were recently used within the high-throughput discovery of a new material for semiconductor spintronics [1], new ferroelectric materials [2] and photovoltaic materials [4].
- W. Chen, J. George, J. B. Varley, G.-M. Rignanese, G. Hautier, “High-throughput computational discovery of In2Mn2O7 as a high Curie temperature ferromagnetic semiconductor for spintronics”, Npj Comput. Mater. 2019, 5, 72.
- M. Markov, L. Alaerts, H. P. C. Miranda, G. Petretto, W. Chen, J. George, E. Bousquet, P. Ghosez, G.-M. Rignanese, G. Hautier, PNAS, 2021, 118, e2026020118
- R. Nelson, C. Ertural, J. George, V. Deringer, G. Hautier, R. Dronskowski, “LOBSTER: Local Orbital Projections, Atomic Charges, and Chemical Bonding Analysis from Projector-Augmented-Wave-Based DFT”, J Comput Chem. 2020, 41, 1931-1940.
- D.F. Dahliah, G. Brunin, J. George, V.A. Ha, G.M. Rignanese, G. Hautier, “High-throughput computational search for high carrier lifetime, defect-tolerant solar absorbers”, Energy Environ. Sci.,, 2021, 14, 5057-5073
Research on Automation in Computational Materials Science
Janine is one of the developers of the high-throughput and data-analysis code pymatgen. She has also contributed to atomate. Janine has recently also written a forum on automation in DFT-based materials science [1].
Bonding Analysis
As already described above, we are expert users of the program Lobster and we have also contributed to the testing and automated usage of this program [1].
Phonons
Our group also focuses on the computation of phonon (vibrational) properties. We have recently contributed to the understanding of low thermal conductivity in Yb14Mn1Sb11 - an important thermoelectric material [1]. We have furthermore helped with the understanding of the relationship between thermal and ionic conductivity [2]. Beyond this, we have also shown that machine-learned interatomic potentials can indeed be used for the computation of accurate phonons for a range of allotropes of Si [3].
- R. Hanus, J. George, M. Wood, A. Bonkowski, Y. Cheng, D. L. Abernathy, M. E. Manley, G. Hautier, G. J. Snyder, R. P. Hermann, Materials Today Physics, 2021, 100344
- T. Bernges, R. Hanus, B. Wankmiller, K. Imasato, S. Lin, M. Ghidiu, M. Gerlitz, M. Peterlechner, S. Graham, G. Hautier, Y. Pei, M. R. Hansen, G. Wilde, G. J. Snyder, J. George, M. Agne, W. Zeier, ChemRxiv 2021, DOI: 10.33774/chemrxiv-2021-3zxh4
- J George, G Hautier, AP Bartók, G Csányi, VL Deringer, “Combining phonon accuracy with high transferability in Gaussian approximation potential models”, J. Chem. Phys., 2020, 153, 044104
Research from Janine’s PhD time on Molecular Crystals:
We recently reviewed our research on molecular crystals: V. L. Deringer, J. George, R. Dronskowski, U. Englert, Acc. Chem. Res., 2017, 50, 1231.
Thermal Motion and Anisotropic Displacement Parameters
Anisotropic displacement parameters (ADPs) describe the mean-squared displacements of atoms in crystals and are typically used within the refinement of diffraction experiments. In recent years, the ab initio calculation of ADPs became available. As my collaborators and I have shown in several recent studies, calculated ADPs from harmonic and quasi-harmonic phonon calculations are in very good agreement with excellent neutron and X-ray diffraction data [1,2,3,4].
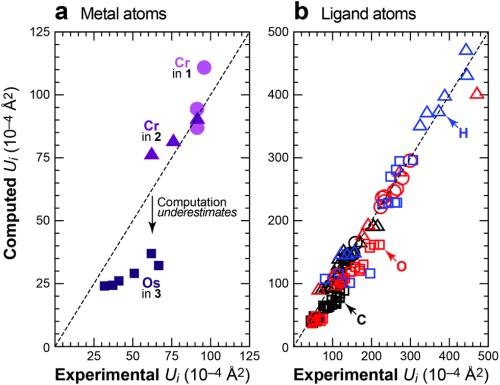
Reprinted from [2] (Dalton Trans. 2016, 45, 13680–13685).
Only very heavy atoms such as osmium still show insufficient results [2]. Especially the ADPs of very light atoms such as hydrogen, that face problems both in X-ray diffraction (e.g., due to contrast problems) and neutron diffraction (e.g., due to incoherent neutron scattering), are in excellent agreement with experimental ADPs [2,4]. You can find more information on this project on www.ellipsoids.de.
- J. George, A. Wang, V. L. Deringer, R. Wang, R. Dronskowski, U. Englert, CrystEngComm 2015, 17, 7414–7422.
- V. L. Deringer, A. Wang, J. George, R. Dronskowski, U. Englert, Dalton Trans. 2016, 45, 13680–13685.
- J. George, V. L. Deringer, A. Wang, P. Müller, U. Englert, R. Dronskowski, J. Chem. Phys. 2016, 145, 234512.
- J. George, R. Wang, U. Englert, R. Dronskowski, J. Chem. Phys. 2017, 147, 074112.
Intermolecular Interactions in Molecular Crystals and Model Systems
Intermolecular interactions such as hydrogen bonds can play a crucial role in the stabilization of crystals. In recent years, also so-called halogen, pnictogen, chalcogen and tetrel bonds were investigated. We assessed the role and importance of hydrogen and halogen bonds for bromomalonic aldehyde [1]. Moreover, we investigated the cooperativity of halogen, chalcogen and pnictogen bonds in infinite chains that are realistic model systems for crystal structures [2].
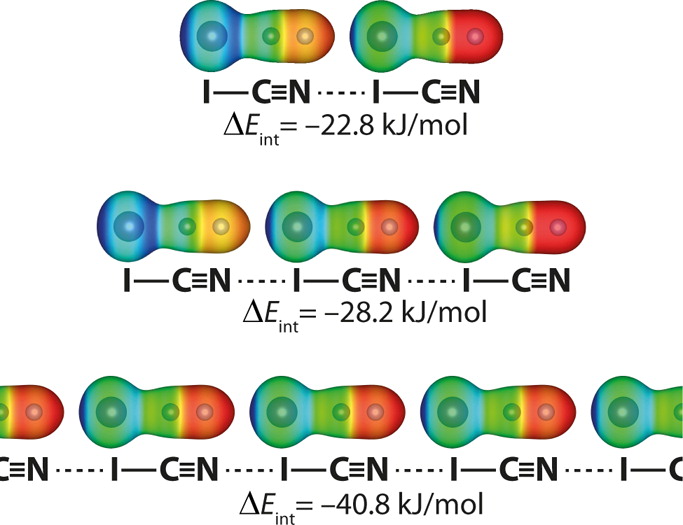
Strengthening of halogen bonds from dimers to chains. Reprinted with permission from J. Phys. Chem. A 2014, 118, 3193–3200. Copyright 2014 American Chemical Society.
- V. L. Deringer, F. Pan, J. George, P. Müller, R. Dronskowski, U. Englert, CrystEngComm 2014, 16, 135–138.
- J. George, V. L. Deringer, R. Dronskowski, J. Phys. Chem. A 2014, 118, 3193–3200.
- J. George, V. L. Deringer, R. Dronskowski, Inorg. Chem. 2015, 54, 956–962.
- J. George, R. Dronskowski, J. Phys. Chem. A 2017, 121, 1381–1387.
Charaterization of Newly Synthesized Molecular Crystals
Several collaborations with experimentalists lead to the successful structural determination and characterization of newly synthesized molecular crystals (guanidinates [1-3], nitride chlorides [4], azides [5], chloro formamidinium compounds [6]). Periodic DFT calculations helped, e.g., with the determination of hydrogen positions and hydrogen ADPs. This is especially relevant for crystals with very heavy atoms (contrast problems in X-ray diffraction or strong absorption in neutron diffraction) and many hydrogen atoms (incoherent neutron scattering). Moreover, the electronic and phononic structures were characterized by periodic DFT calculations.
- R. Missong, J. George, A. Houben, M. Hoelzel, R. Dronskowski, Angew. Chem. Int. Ed. 2015, 54, 12171–12175.
- A. L. Görne, J. George, J. van Leusen, G. Dück, P. Jacobs, N. K. C. Muniraju, R. Dronskowski, Inorg. Chem. 2016, 55, 6161–6168.
- A. Görne, J. George, J. van Leusen, R. Dronskowski, Inorganics, 2017, 5, 10.
- X. Liu, J. George, S. Maintz, R. Dronskowski, Angew. Chem. Int. Ed. 2015, 54, 1954–1959.
- Y. Li, J. George, X. Liu, R. Dronskowski, Z. Anorg. Allg. Chem. 2015, 641, 266–269.
- A. Möller, J. George, R. Dronskowski, Z. Anorg. Allg. Chem. 2018, 644, 1485.